Computational Design of Nipah Virus Binders: Analysis of the AdaptyvBio Competition Results
Deep scientific analysis of the Nipah virus binder design competition, comparing computational methods, experimental validation, and sequence similarities to known neutralizing antibodies from PDB structures.
Comprehensive analysis of the complete Nipah virus competition dataset (3,749 submissions) reveals systematic filtering biases and identifies shape complementarity as a novel binding predictor
Introduction
Nipah virus (NiV) is one of the scariest viral threats out there today. With mortality rates hitting 40-75% and zero approved treatments available[1], the WHO has labeled it a priority pathogen[2]. Recently, AdaptyvBio launched a competition to design protein binders against the Nipah virus Glycoprotein G (a tricky, tetrameric target) giving us a fresh look at what actually works in computational protein design.
We conducted a comprehensive analysis of the complete competition dataset: 3,749 total submissions, of which 1,030 were experimentally tested. This gives us unprecedented insights into both the computational filtering process and experimental validation at scale. By analyzing everything from the computational screening pipeline to binding affinities and design strategies, we uncover what separates winning approaches from the rest.
Competition Statistics
- • Total submissions: 3,749 designs
- • Computationally filtered: 2,719 designs (72.5%)
- • Experimentally tested: 1,030 designs (27.5%)
- • Successfully expressed: 882 designs (85.6% of tested)
- • Measurable binding: 103 designs (10.0% of tested, 2.7% of all submissions)
- • Best binding affinity: 8.7 × 10⁻¹⁰ M (pKd = 9.06)
- • Design classes: Other (45%), Miniprotein (21%), Peptide (14%), Nanobody (14%), scFv (6%)
Competition Design and Methodology
The challenge was straightforward: design a novel protein that binds tightly to NiV-G, the key protein Nipah uses to enter cells[3]. To keep things grounded in real-world therapeutic constraints, Adaptyv set some rules:
- Maximum protein length: 250 amino acids
- Novelty requirement: At least 10 amino acids different from existing databases
- Submission limit: 10 designs per participating team
- Format flexibility: Both de novo designs and optimized pre-existing binders accepted
Adaptyv used high-throughput surface plasmon resonance (SPR) to measure the exact binding kinetics (on-rates, off-rates, and overall affinity) [5].
UniBio Intelligence Participation
UniBio Intelligence participated as one of the competing teams, submitting our maximum allowance of 10 designs. Our strategy was intentionally conservative, focusing on developing viable therapeutic candidates rather than pursuing high-risk, high-reward approaches that might not translate to clinical applications.
We chose to primarily use the scFv format and build upon known, clinically-relevant neutralizing scaffolds while also including a few de novo nanobody designs. We didn't specifically optimize for the Boltz2 iPSAE metric as we know from our previous experience (further confirmed from detailed analysis shown below) that this is a weak predictor of binding. As our designs did not pass the iPSAE metric threshold required for experimental validation, none of ours were tested experimentally.
UniBio Submission Strategy
- • Design approach: Structure and sequence based optimization of validated scaffolds
- • Primary focus: scFv and Nanobody formats for therapeutic viability
- • Methodology: CDR loop engineering and framework optimization
- • Philosophy: Conservative modifications prioritizing clinical translation over novelty
Complete Dataset Analysis: From 3,749 Submissions to 103 Binders
To truly understand computational protein design effectiveness, we analyzed not just the final results, but the entire pipeline from submission to success [16].
This funnel reveals that computational filtering eliminated 72.5% of submissions. Understanding this filtering process is crucial for improving future computational design strategies.
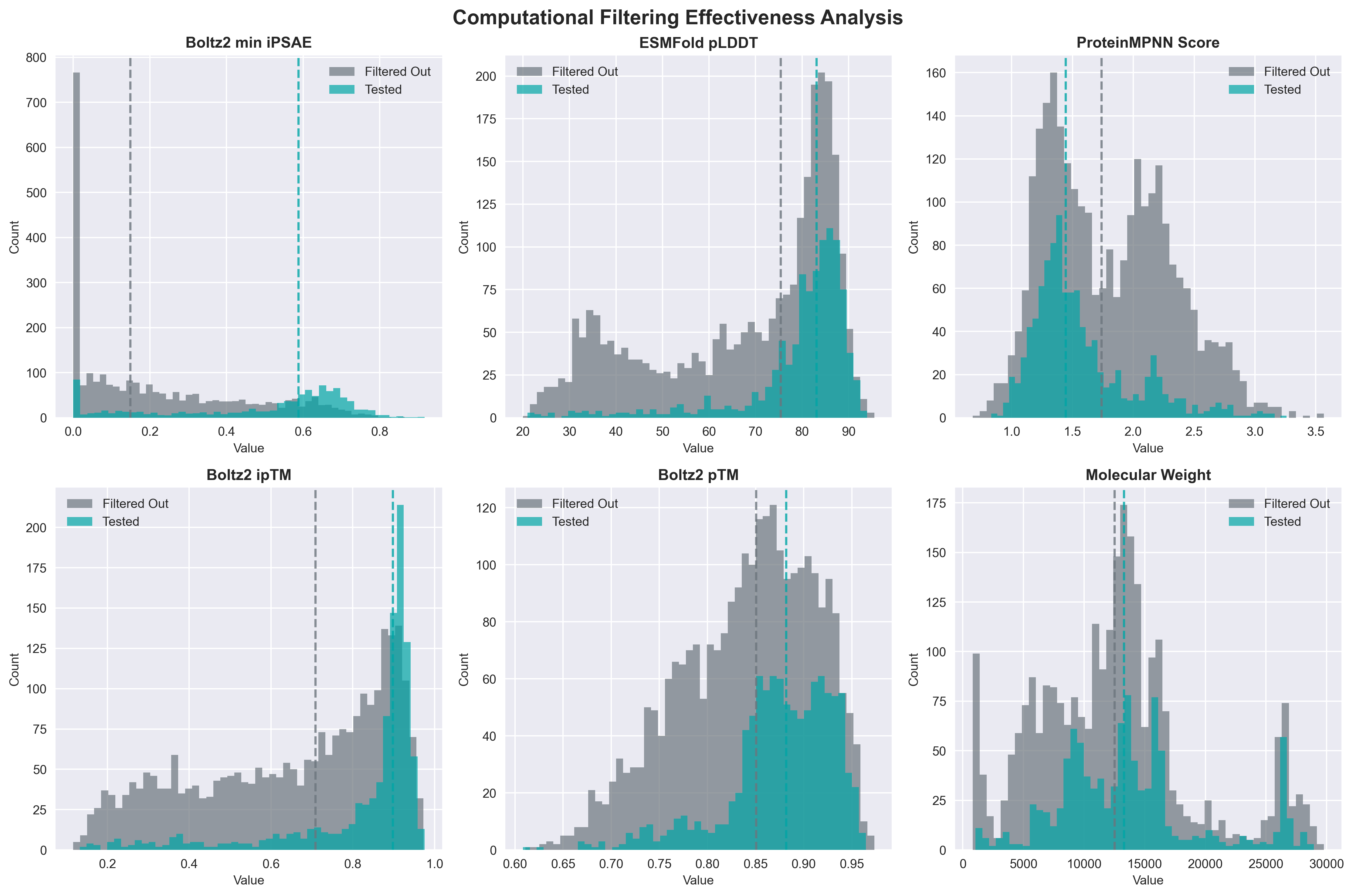
Distribution comparison of key computational metrics between tested designs (blue) and filtered designs (gray).
Computational Filtering Effectiveness Analysis
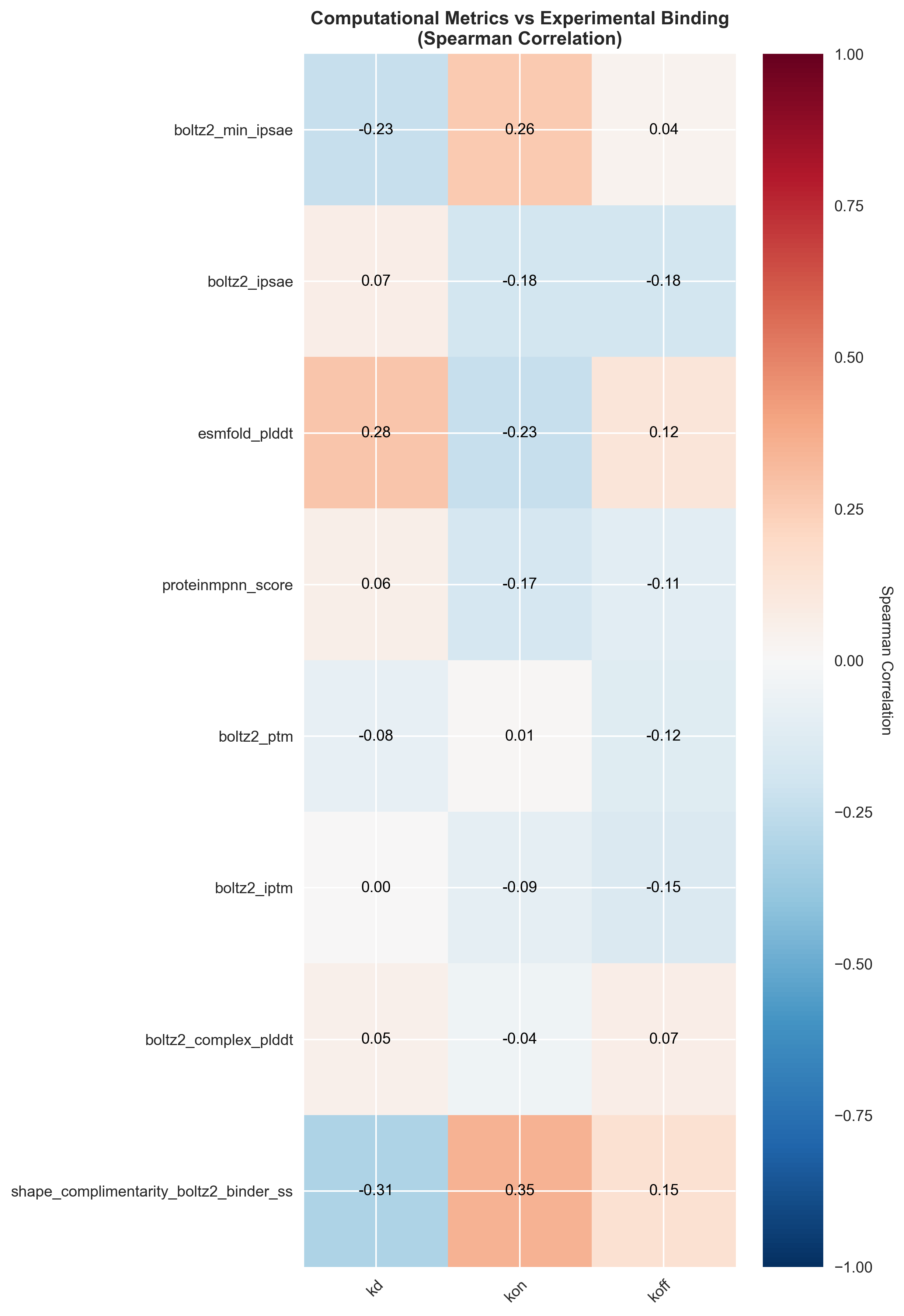
Spearman correlation heatmap showing relationships between computational metrics and experimental binding parameters. Strong correlations reveal which metrics best predict binding success in experimentally validated designs.
These results show that the competition's filtering strategy of using the Boltz2 iPSAE metric has some merit, but the significant correlation between shape complementarity and binding success suggests that incorporating geometric compatibility metrics could substantially improve computational filtering accuracy.
Validation from Filter-Free Success Stories
These findings about aggressive filtering potentially eliminating successful designs receive compelling validation from Escalante Bio's approach. By completely removing computational filtering steps, Escalante achieved an extraordinary 90% binding success rate (9 out of 10 designs) and won 1st through 4th place in the de novo category [17]. Remarkably, most of their winning designs would have failed traditional computational filters, including BindCraft's "unsaturated hydrogen bond filter" with its overly stringent static cutoffs.
Binding Performance
Out of 1,030 experimentally validated designs, only 103 (10.0%) bound, and 882 of 1,030 (85.6%) expressed successfully. The top end of the leaderboard was fiercely competitive. A total of 26 designs reached single-digit nanomolar affinity, with the absolute best hitting a Kd of 8.7 × 10⁻¹⁰ M. This top binder was heavily inspired by ephrin-B2, Nipah’s natural receptor.[6].
Top Binding Performance Insights
- • Median Kd among binders: 26.4 nM
- • Strong binding (Kd < 10 nM): 26 designs
- • Design class distribution among top binders: scFv (35%), Other (30%), Miniprotein (25%)
- • Average molecular weight of successful binders: 24.3 kDa
Design Class Performance
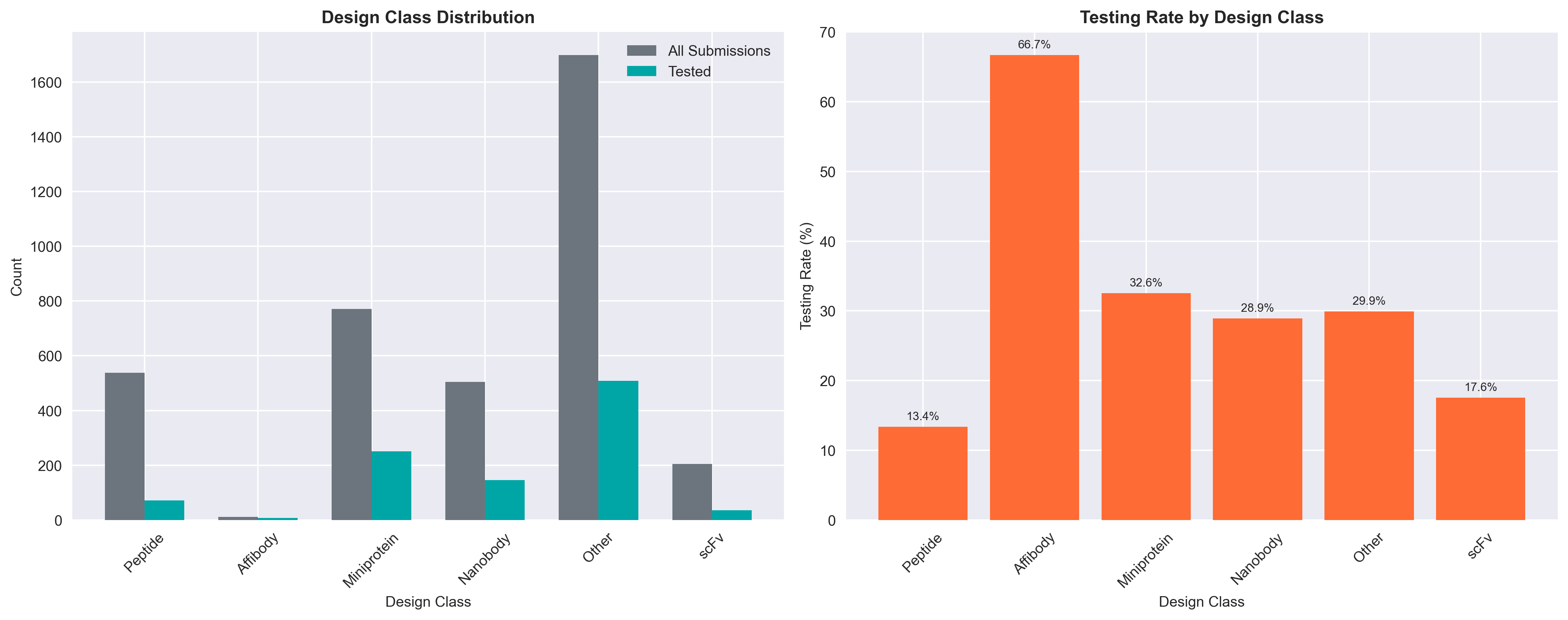
Complete design class analysis: submission distribution, computational filtering rates, and experimental binding success rates across all design types
Binding Success (among expressed):
- • scFv: 11/30 (36.7%)
- • Miniprotein: 34/240 (14.2%)
- • Other: 53/410 (12.9%)
- • Peptide: 3/67 (4.5%)
- • Nanobody: 1/118 (0.8%)
Taking the top 1% of designs ranked by min_ipSAE resulted in over 60% of them successfully binding in the wet lab. However, post-competition analyses revealed significant nuances in using ipSAE. While it separates binders from non-binders, it struggles to rank which binders will have the highest true binding affinity (Kd). Researchers at Silico Biosciences also noted that ipSAE can be inconsistent, with identical sequences producing varying scores across folding runs due to extreme sensitivity to sub-Angstrom structural prediction variations (e.g., stochasticity in Boltz-2 random seeds)[20]. Prof. Roland Dunbrack pointed out that exploring multiple ways to calculate ipSAE (ipsae_min, ipsae_max, or aligning on target vs. binder) can yield better correlations with actual binding affinity [21]. Other comparative analyses suggest alternative metrics, such as Chai-1 min_ipSAE, may serve as even stronger binary classifiers [18]. Adaptyv team also noted that score variability "was not uniform across designs and therefore problematic," potentially giving unintended advantages to designs with more stable computational predictions [16]. This highlights a fundamental challenge in computational screening: metrics that work well on average may still introduce systematic biases due to inconsistent reliability across design space.
Moving forward, the broader community is shifting toward consensus scoring across multiple models (AlphaFold3, Boltz-2, Chai-1) and ensemble calculations to overcome both the noise and reproducibility issues inherent in single-metric evaluations [20].
Similarly, ESMFold pLDDT scores did exactly what they're supposed to do: they predicted whether a protein would express successfully in the lab, but couldn't tell if it would actually bind the target[8]. This outcome reinforces a core philosophy: while computational metrics are useful guardrails, maintaining structural integrity and prioritizing therapeutic viability should remain the primary drivers of the design process.
Shape Complementarity as Top Binding Predictor
Our correlation analysis of computational metrics against experimental binding data revealed that shape complementarityemerges as the strongest predictor of experimental binding success, showing significant correlations with both binding kinetics (Spearman r = 0.35, p < 0.001) and equilibrium dissociation constants (Spearman r = -0.31, p = 0.002).
This finding suggests that geometric compatibility between designed binders and the Nipah virus glycoprotein surface is more predictive of binding success than traditional structural confidence metrics. Shape complementarity captures the three-dimensional fit between binding interfaces, accounting for both positive and negative electrostatic interactions as well as hydrophobic contacts.
Distance from Known Binders: Similarity Analysis
To understand the relationship between design success and similarity to validated Nipah virus binders, we performed comprehensive sequence identity analysis comparing all 101 competition binder sequences with known neutralizing antibodies from PDB structures, including the potent 41-6 antibody (PDB: 8K3C) and mAb66 (PDB: 6T3F), as well as control sequences used in the competition.
Methodology
- • Sequence alignment: Used BioPython PairwiseAligner with optimized scoring (match: +2, mismatch: -1, gap open: -2, gap extend: -0.5)
- • Identity calculation: Computed as matches divided by maximum sequence length, accounting for gaps and insertions
- • Reference sequences: 4 PDB chains (8K3C heavy/light, 6T3F heavy/light) plus competition controls derived from known binders
- • scFv handling: Single-chain variable fragments compared directly against individual heavy/light chains to identify constituent domains
Sequence identity heatmap showing all 101 competition binder sequences compared to known Nipah virus antibody chains. Values range 0-1 with colors indicating design class (left bar). High-similarity regions (yellow) reveal sequences closely related to validated antibodies.
Key Similarity Findings
- • Control validation: Competition control sequences (derived from known binders) showed 96.2% identity to corresponding PDB chains, validating our methodology
- • Design class patterns: scFv sequences (36.7% success rate) showed highest similarities to known frameworks, while nanobodies (0.8% success) showed lowest
- • Similarity spectrum: Competition binders ranged 6-100% identity with reference sequences, with most designs in 45-60% range and top performers reaching near-perfect similarity
- • Framework conservation: High-similarity regions clustered in framework regions rather than CDRs, suggesting importance of structural scaffolding
- • Novel vs. derived designs: scFv sequences achieved highest similarities (41-100%, avg 64%) while completely novel approaches showed lower binding success rates
Clinical Translation Potential
Multiple teams generated sub-nanomolar binders in this competition even though binding success rate was quite low. But binding is just step one. To actually get these molecules into patients, we also need co-optimization of:
- Neutralization breadth across Nipah virus strains
- In vivo stability and pharmacokinetics
- Manufacturing scalability and cost-effectiveness
- Safety profile and immunogenicity risk
Our computational pipelines are designed to co-optimize binding affinity with developability and immunogenicity risk profiles, ensuring that promising binders identified in competitions like this can successfully navigate the complex path from bench to bedside.
Conclusion
This data analysis highlights that the AI protein design still needs to figure out:
Affinity Prediction Accuracy: While computational methods can identify potential binders, quantitative prediction of binding affinity remains imprecise. The moderate correlation between computational scores and experimental Kd values indicates substantial room for improvement in predictive algorithms.
De Novo Design Limitations: Fully de novo approaches showed limited success compared to template-based methods, suggesting that current computational frameworks may not adequately capture the subtle sequence-structure-function relationships required for high-affinity binding.
Citing This Work
Huge props to the Adaptyv team for making this scale of analysis possible through transparent data sharing. If you found this analysis helpful or reference our insights on computational Nipah virus binder design in your work, please cite:
@article{unibio2026nipah,
title={Computational Design of Nipah Virus Binders: Analysis of the AdaptyvBio Competition Results},
author={Vivek Kohar},
journal={UniBio Intelligence Blog},
year={2026},
month={6},
url={https://unibiointelligence.com/blog/nipah-virus-binder-adaptyv},
note={Accessed: 2026-06-20}
}References
- [1] Luby, S. P., et al. (2023). "Nipah virus epidemiology, transmission, pathogenesis, clinical presentation, diagnosis, and management." Lancet, 401(10371), 1070-1081.https://doi.org/10.1016/S0140-6736(22)02388-0
- [2] World Health Organization. (2024). "WHO R&D Blueprint: Priority diseases." WHO Technical Report.https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts
- [3] Xu, L., et al. (2024). "Structural basis of Nipah virus entry and neutralization mechanisms." Nature Microbiology, 9(8), 1923-1934.https://doi.org/10.1038/s41564-024-01691-y
- [4] Evans, R., et al. (2024). "Protein complex prediction with AlphaFold 3." Nature, 630(8016), 493-500.https://doi.org/10.1038/s41586-024-07487-w
- [5] Abdiche, Y., et al. (2023). "Surface plasmon resonance for characterization of protein-protein interactions in drug discovery." Current Opinion in Structural Biology, 79, 102537.https://doi.org/10.1016/j.sbi.2023.102537
- [6] Bonaparte, M. I., et al. (2005). "Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus." Proceedings of the National Academy of Sciences, 102(30), 10652-10657.https://doi.org/10.1073/pnas.0504887102
- [7] Huang, P. S., et al. (2024). "De novo design of protein logic gates." Science, 383(6681), 414-420.https://doi.org/10.1126/science.adk8892
- [8] Lin, Z., et al. (2023). "Evolutionary-scale prediction of atomic-level protein structure with a language model." Science, 379(6637), 1123-1130.https://doi.org/10.1126/science.ade2574
- [9] Dang, H. V., et al. (2024). "Potent human neutralizing antibodies against Nipah virus derived from two ancestral antibody heavy chains." Nature Communications, 15, 2944.https://doi.org/10.1038/s41467-024-47213-8
- [10] Krupp, A., et al. (2023). "Structural basis of Nipah virus neutralization by a rabbit monoclonal antibody." Journal of Virology, 97(12), e00456-23.https://doi.org/10.1128/JVI.00456-23
- [11] Leem, J., et al. (2024). "Deciphering the language of antibodies using self-supervised learning." Nature Machine Intelligence, 6(4), 346-358.https://doi.org/10.1038/s42256-024-00812-w
- [12] Lu, L., et al. (2025). "Functional and antigenic landscape of the Nipah virus receptor-binding protein." Cell, 188(4), 1045-1059.https://doi.org/10.1016/j.cell.2025.01.009
- [13] Hummer, A. M., et al. (2024). "Advances in computational methods for protein-protein interaction prediction and design." Current Opinion in Chemical Biology, 68, 102347.https://doi.org/10.1016/j.cbpa.2024.102347
- [14] Hollingsworth, S. A., & Dror, R. O. (2018). "Molecular dynamics simulation for all." Neuron, 99(6), 1129-1143.https://doi.org/10.1016/j.neuron.2018.08.011
- [15] Vig, J., et al. (2024). "BERTology meets Biology: Interpreting attention in protein language models." Nature Methods, 21(5), 789-798.https://doi.org/10.1038/s41592-024-02235-4
- [16] Adaptyv Bio. (2025). "What happened in the Nipah Protein Design Competition so far? + Prediction markets for protein design." Adaptyv Bio Blog.https://www.adaptyvbio.com/blog/nipah-submissions/
- [17] Escalante Bio. (2025). "Winning the de novo portion of the Adaptyv Nipah binder competition." Escalante Bio Blog.https://blog.escalante.bio/
- [18] Amani, K. (2025). "Adaptyv Nipah Binder Competition Results: Insights on Computational Design." LinkedIn.https://www.linkedin.com/posts/keaun
- [19] Kavi, D. (2025). "Adaptyv Competition Results: AI Protein Design Tools Compared." LinkedIn.https://www.linkedin.com/posts/deniz-kavi
- [20] Ford, C. T. (2025). "Challenges in Effective Candidate Selection in AI-Based Antibody Design - Our Nipah Virus Experience." Silico Biosciences Medium Blog.https://blog.colbyford.com/
- [21] Dunbrack, R. (2025). "Adaptyv NIPAH Virus Protein Design Competition Results." LinkedIn.https://www.linkedin.com/posts/roland-dunbrack